Research
I am interested in gaining insight into physico-chemical systems using simulation and computations.
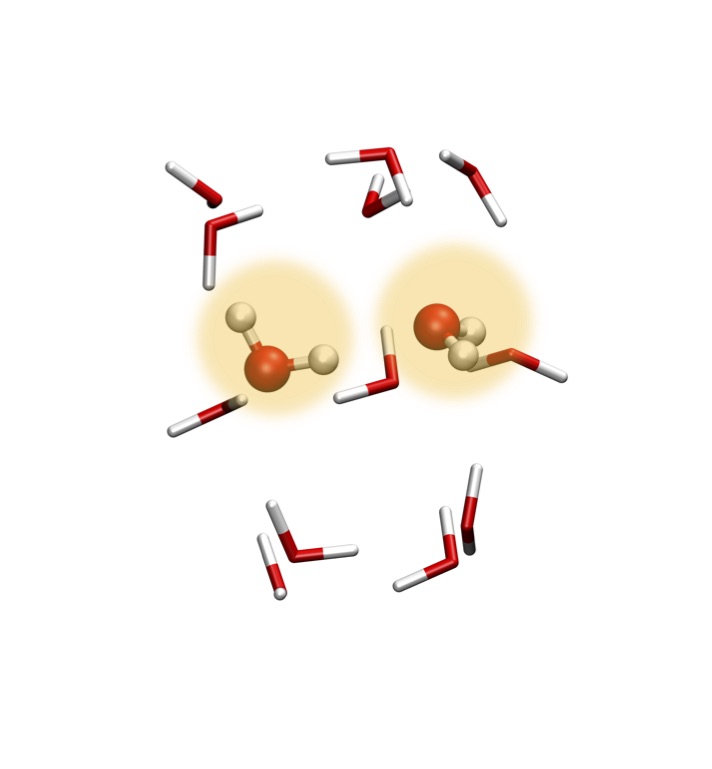
Along with Dr. Joonho Lee, we developed an embedded many-body expansion method for mean-field systems called the Polarized many-Body Expansion (PolBE). This method takes advantage of the locality of chemical interaction to devise a technique to compute the properties of molecular clusters at O(N2) cost instead of the traditional O(N3). This method is also can also take advantage of modern day high-performance computing architectures by parallelizing efficiently over any number of CPU cores. This technique has been implemented in C++ using Armadillo in Q-Chem .
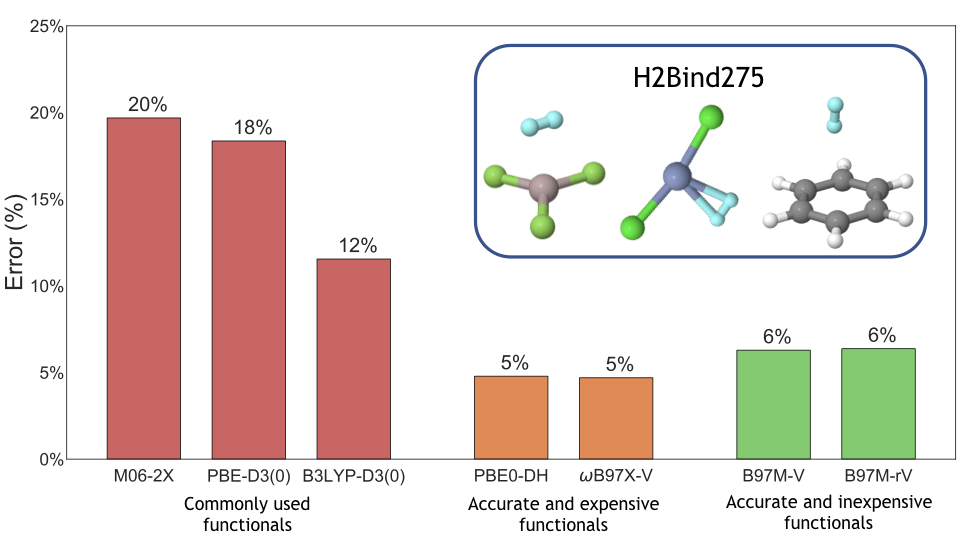
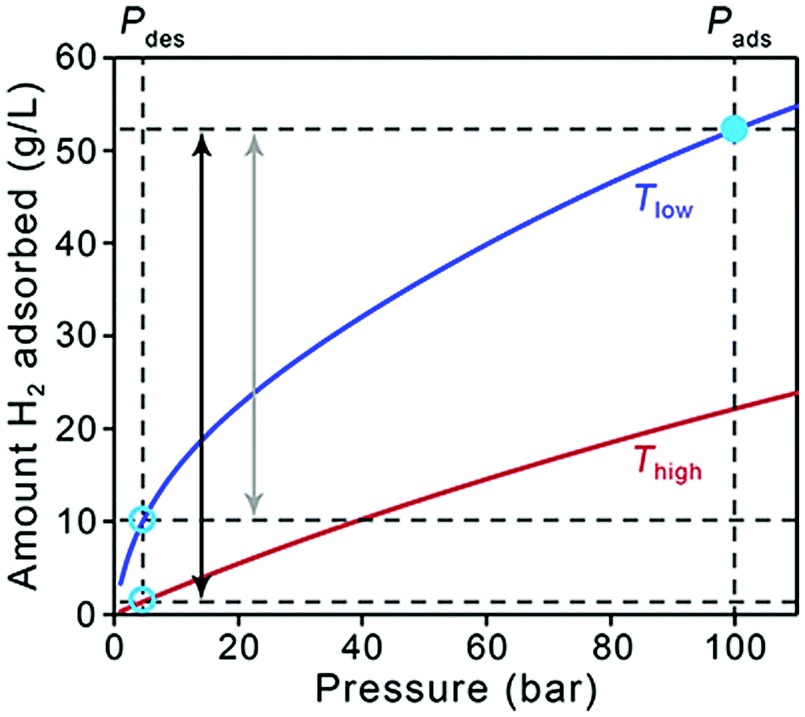
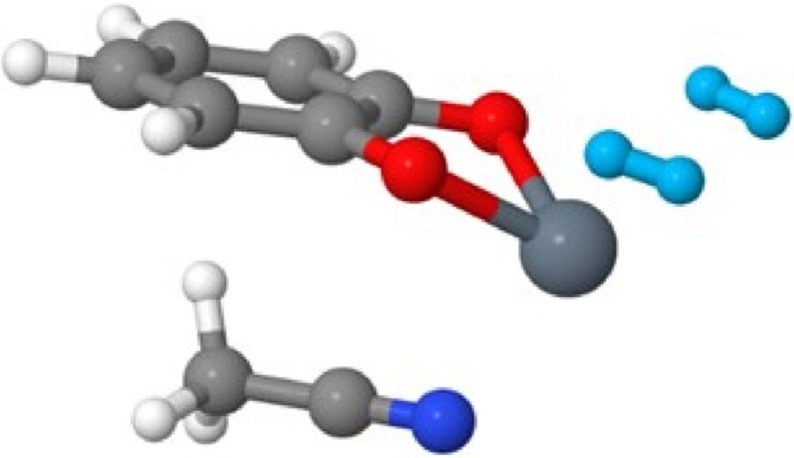
I have also worked on various aspects of identification and assessment of materials for on-board vehicular hydrogen storage as a part of the Department of Energy’s Hydrogen Materials – Advanced Research Consortium ( HyMARC ). I have used computational methods to investigate metalated catecholates for adsorbing multiple hydrogens at a single site. Recently, I have comprehensively assessed the performance of 55 density functionals for hydrogen storage. For this purpose, I implemented an automated workflow in python using pandas for the end-to-end management of more than 20,000 data points with different metadata structures.
Recently, I explored an idea to predict protein function from protein sequence information using machine learning techniques. In this work, we used the sequence-based deep learning representation of protein sequences called UniRep to predict different properties of the protein like protein function, location of active sites, and role of active sites. Inspired multi-scale modeling in physics and chemistry, we used two different levels of representation: one for the protein sequence (UniRep) and another for the amino acid residue (message passing neural network based link to). The proposed model can identify the role of active site amino acids with 88% accuracy. This model was implemented in python using NumPy and TensorFlow. The implementation can be found on my Github .